Welcome to BML
BML is an online tool that allows the user to discover and analyze the results of Motifs quickly. BML utilizes both position weight matrix (PWM) and dinucleotide weight matrix (DWM), the latter of which enables the expression of the interdependencies of neighboring bases. Users access two methods for motifs discovery from sequences data, the first Classic way that users must set some parameters, and the second users use of Parameter-Free (PF) way that does not need to set parameters.
Most motif discovery web tools push certain levels of burden for motif parameterization to non-expert users, such as the motif length, direction, gap range, the number of occurrences, or the number of genes that include motifs. Some tools attempt to circumvent this problem by setting reasonable (yet not optimized) default parameters, however leading to sub-optimal performance.
We to lessen the burden of motif parameterization for users but still deliver optimal performance, proposes using BML-PF.
In addition, BML can just show the Web-Sequence logo and heatmap if users can access real motifs sequences (PWM and DWM).
Getting Run:
Input Data:
For input sequence format, you have two choices: Text Format or Fasta Format, and BML supports both DNA and RNA sequences. As default, BML is set on Fasta format and DNA sequence, but you can change it according to your input data.

BML has two main methods to find motifs, first the classic BML way that users must set parameters, second BML-PF that users do not need to set any parameters to motifs discovery. In each method, BML can process by PWM (Position Weight Matrix) or DWM (Di-nucleotide Weight Matrix).
According to the classic method, users must set these parameters:

Left motif size (bp)
Right motif size (bp)
Minimum gap between two block motifs (bp)
Maximum gap between two block motifs (bp)
Note: BML is searching two-block motifs in each site as default. If you want to find one block motif in each site, just set 0 value for the right motif size, minimum gap, and maximum gap parameters.
Number of times trying to repeat the process to find the best motif (default 30)
Checking this box instructs BML to check the reverse complement of the input sequences for motif sites.
Select the site distribution
This is where you tell MEME how you believe occurrences of the motifs are distributed among the sequences. Selecting the correct type of distribution improves the sensitivity and quality of the motif search.
* Zero or One Occurrence per Sequence (zoops) MEME assumes that each sequence may contain at most one occurrence of each motif. This option is useful when you suspect that some motifs may be missing from some of the sequences. In that case, the motifs found will be more accurate than using the One Occurrence per Sequence option. This option takes more computer time than the One Occurrence Per Sequence option (about twice as much) and is slightly less sensitive to weak motifs present in all of the sequences.
* One Occurrence Per Sequence (oops) MEME assumes that each sequence in the dataset contains exactly one occurrence of each motif. This option is the fastest and most sensitive but the motifs returned by MEME will be "blurry" (less specific) if they do not occur in every input sequence.
* Any Number of Repetitions (anr) MEME assumes each sequence may contain any number of non-overlapping occurrences of each motif. This option is useful when you suspect that motifs repeat multiple times within a single sequence. In that case, the motifs found will be much more accurate than using one of the other options. This option can also be used to discover repeats within a single sequence. This option takes much more computer time than the One Occurrence Per Sequence option (about ten times as much) and is somewhat less sensitive to weak motifs that do not repeat within a single sequence than the other two options.
Result:
Sequence web logo

A sequence logo is created from a collection of aligned sequences and depicts the consensus sequence and diversity of the sequences.
Heatmap
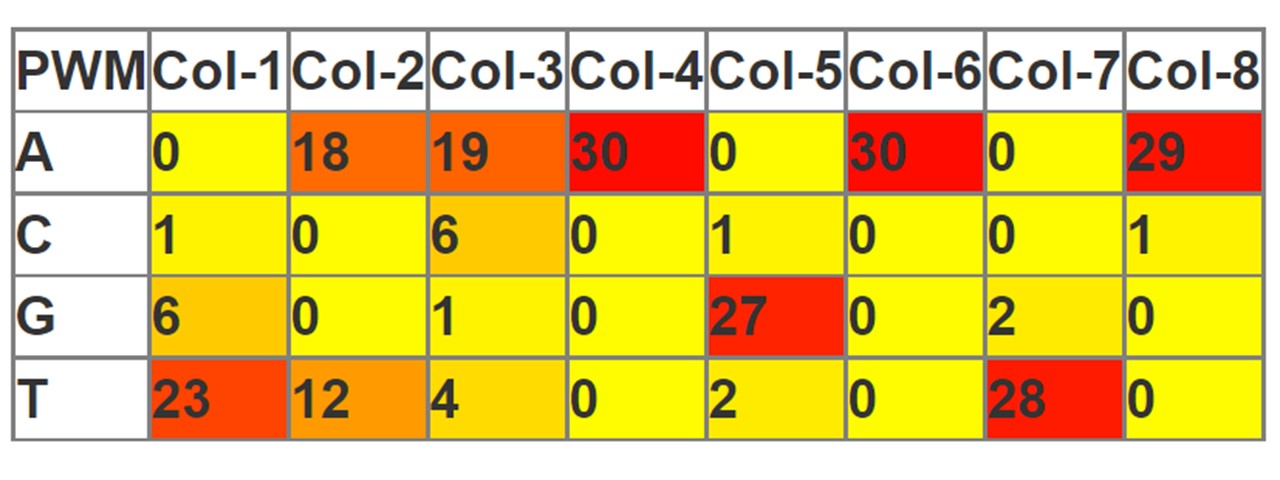
A position weight matrix (PWM), or Dinucleotide weight matrix (DWM), is a commonly used representation of motifs (patterns) in sequences.
Text format result:
* Entropy score for each process to find motif
* Left/Right motif size
* Start position of Left/Right motif
* Position Weight Matrix
* Sequence motifs
Download
After "Run" of BML for download your results, just click on "Download" button.
If you used this tool for your work, please cite: "Vahed, Mohammad, Majid Vahed, and Lana X. Garmire. "BML: a versatile web server for bipartite motif discovery." Briefings in Bioinformatics 23, no. 1 (2022): bbab536."